Sickle Cell Disease: Essential Information for Africans
By: Tii Ngwachi Munghieng, MD. Medically reviewed by A. Odutola; MBBS, PhD, FRCSEd.
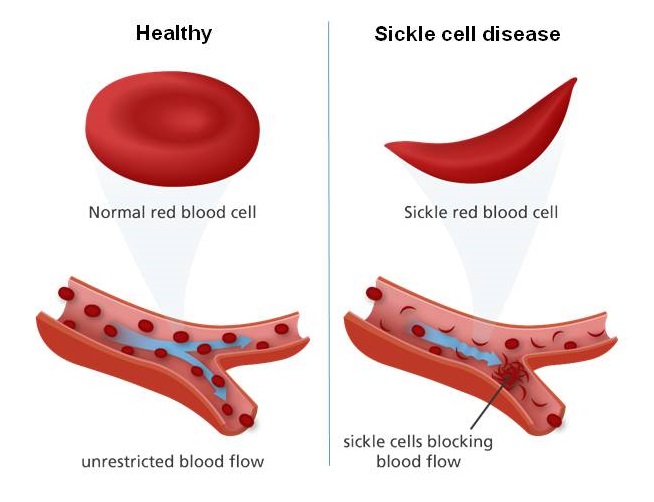
Comparison of normal round red blood cells with crescent-shaped sickle cells, which block blood vessels in sickle cell disease. Click on image toenlarge
Introduction
At least 240,000 children in Africa are born annually with sickle cell disease (SCD). [1] SCD is a common genetic blood disorder marked by red blood cells that take on an abnormal crescent (half-moon) or sickle shape. This impairs their ability to travel normally through blood vessels. Among the various forms of SCD, sickle cell anemia (SCA) is the most severe form.
The Sickle Cell Trait (SCT) plays a central role in the inheritance of SCD. SCT occurs when a person inherits one normal hemoglobin gene (HbA) and one sickle hemoglobin gene (HbS), typically causing no symptoms. However, when two individuals with SCT have children, there is a 25% chance the child will inherit SCD. This genetic mechanism contributes significantly to the high prevalence of SCD in Africa.
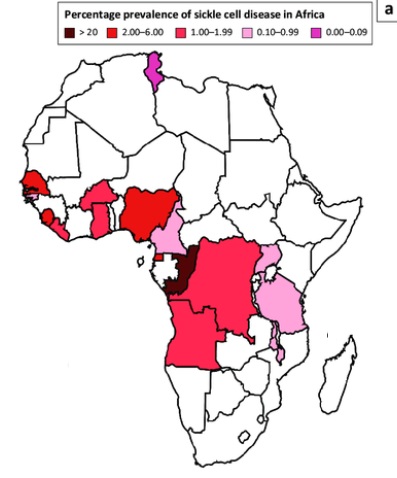
Fig. 1: Percentage prevalence of sickle cell disease in Africa. Image credit: Twum et al. 2023. Click on image to enlarge.
In many African countries, especially those in Central and West Africa (see figure 1) like Cameroon, Ghana, and Nigeria, SCD prevalence ranges from 20% to 30%, and in some regions of Uganda, it can reach as high as 45%. [2] Tragically, most children with severe forms of the disease die before the age of five.
This article explains why SCD is so common in Africans, examining the role of genetics, the sickle cell trait (SCT), evolutionary advantages, and the link to malaria.
.jpg)
Burden of sickle cell disease in Africa, Click on image to enlarge.
Africa bears the heaviest burden of Sickle Cell Disease (SCD) globally. Worldwide, an estimated 515,000 children are born with sickle cell disease every year, and nearly 80% occur in sub-Saharan Africa. 50-80% of people with SCD die before the age of five in some regions due to lack of diagnosis and treatment [3].
SCD has no gender selection. It affects males and females equally, as it is inherited in an autosomal recessive manner, meaning the condition is not linked to sex chromosomes [4].
Sickle Cell Trait (SCT), the carrier state (HbAS), affects 15-30% of the population in sub-Saharan Africa [4]. SCT offers some protection against severe malaria, which explains its high prevalence in regions where malaria is endemic.
Although the sickle cell trait (SCT) is protective against malaria, when two partners with SCT give birth to a child, there's theoretically a 1 in 4 (25%) chance of the child having SCD. This occurs when an individual inherits two sickle cell genes (HbS)—one from each parent. These abnormal genes produce defective hemoglobin (oxygen-carrying unit in blood) called hemoglobin S. Under low oxygen conditions, this hemoglobin causes red blood cells to deform into a sickle shape, making them rigid and sticky [4].
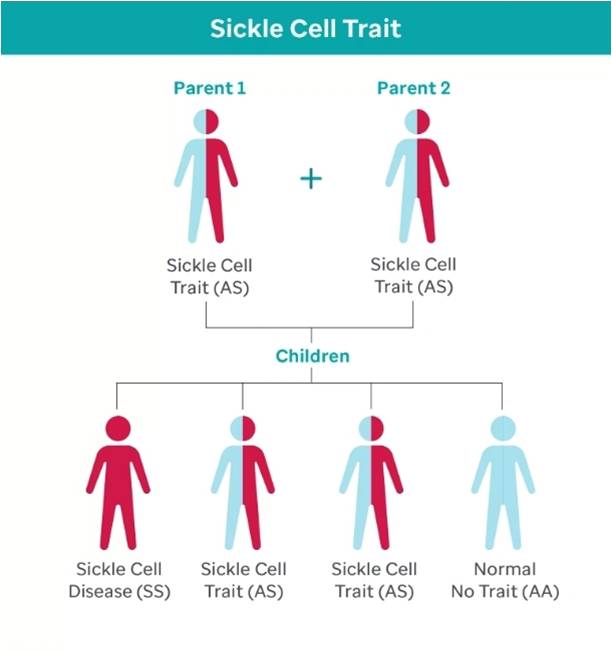
Sickle cell inheritance diagram showing a 1 in 4 chance of sickle cell disease (SS) when both parents have the sickle cell trait (AS). Click on image to enlarge.
The sickled cells:
Due to the protective role of SCT against malaria, the survival rate from severe malaria is high, leaving a high probability of producing children with SCD. But SCD negatively impacts Africans in the following ways;
All of these cause a decline in the value of goods and services produced (GDP) in Africa in any given period.
SCD can be recognized or diagnosed through many signs and symptoms.
Sickle Cell Disease usually becomes apparent after 6 months of age, as fetal hemoglobin (HbF), which protects against symptoms, decreases [5],
Common symptoms and signs include:

Infographics on common symptoms of sickle cell disease. Credit.
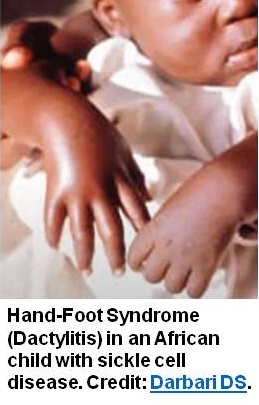
SCD can be diagnosed when someone presents to a healthcare professional with complaints like bone and joint pains, pale appearance, fatigue, etc.
The healthcare professional will take a detailed medical history, perform a physical examination and depending on the circumstances, may order some of the following tests to confirm a clinical diagnosis:
Other methods of SCD diagnosis like Newborn screening and genetic testing are gradually gaining ground in Africa.
While there is no universal cure for SCD, several treatments options can help patients live healthier lives. These include:
Below are simple explanations of how SCD is treated.
a) Pain Relief
Pain crises are a common complication of SCD. Treatment includes:
b) Hydroxyurea
c) Antibiotics
d) Folic Acid Supplements
e) Crizanlizumab
Patients with SCD can manage their condition by adopting healthy habits and working closely with healthcare providers. Self care options include:
a) Staying Hydrated
Drinking plenty of water prevents dehydration, which can trigger pain crises.
b) Eating Healthy Diet
Eating a balanced diet with adequate vitamins and minerals, including folic acid, supports red blood cell production.
c) Prevent Infections
Vaccinations, including the pneumococcal and meningococcal vaccines, protect against severe infections.
Prompt treatment with antibiotics is crucial when infections occur.
d) Regular Check-ups
Routine medical visits help monitor the disease, prevent complications, and ensure treatments are effective.
Living with SCD can be emotionally and socially challenging. Psychological and social support include:
This treatment is risky and not widely available in many parts of the world, especially in Africa. It is most successful in children with severe SCD who have a closely matched sibling donor.
Sickle cell disease can cause a host of complications due to organ damage, infections and blood clots. The complications can be grouped into acute or sudden, chronic or late and those associated with pregnancy as detailed in Table 1.
.jpg)
Table 1: Showing some common complications of sickle cell disease. Click on image to enlarge.
The processes underlying the various complications can be traced to the poor oxygen carrying ability of sickled red blood cells, their easy destruction, and their ability to stick to blood vessels and block them.
While the sickle cell disease cannot be cured in most cases, it can be prevented by addressing its genetic transmission. Some preventive measures are outlined below;
Screening and counseling are vital for preventing SCD in Africa, where premarital and prenatal genetic testing is often unavailable.
Many African communities have limited awareness about SCD and its genetic nature. Addressing this gap is crucial:
For couples identified as carriers, there are several options to prevent having a child with SCD. These include:
Though expensive and not widely accessible in Africa, this method ensures that embryos without SCD are conceived.
Couples can choose to use sperm or eggs from donors who do not carry the sickle cell gene.
Adoption offers an alternative for couples who want to avoid the risk of passing on SCD.
While newborn screening does not prevent SCD, it ensures early diagnosis, which can reduce complications and improve quality of life with early quality care. Many African countries still lack widespread newborn screening programs [7].
SCD in Africa poses a lot of challenges both for patients, families, and healthcare systems. Some of these challenges are outlined as follows;
Africa has the highest global burden of SCD, with nearly 80% of the world’s cases occurring on the continent. Many children are born with the disease, but due to limited awareness and diagnosis, most remain untreated.
Many people in African communities lack knowledge about SCD, its causes, and management. Stigma and misconceptions, such as associating SCD with curses or witchcraft, often prevent patients from seeking care.
Newborn screening is rare in most African countries, leading to late diagnosis of SCD. Without early detection, children are more likely to suffer severe complications or die before the age of five.
Healthcare systems in many African countries face challenges such as:
The cost of managing SCD is often unaffordable for families, especially in low-income settings. This includes the cost of medications, hospital visits, and blood transfusions, which are vital for managing the disease.
SCD causes many health problems such as pain crises, severe anemia, infections (due to a weakened immune system), stroke, etc. Due to the limited healthcare resources in Africa, these complications can result in disability or death.
Living with SCD often causes emotional and social difficulties. Patients often face stigma, discrimination, and isolation. Families also struggle with the emotional burden of caring for a child with a chronic disease.
SCD research and policy development remain underfunded in Africa. This lack of attention limits progress in improving patient care, raising awareness, and supporting families.
Sickle Cell Disease (SCD) continues to be a major health concern in Africa, affecting millions of individuals. While efforts to improve awareness, diagnosis, and treatment are ongoing, challenges such as limited healthcare infrastructure, high costs, and stigma persist. To improve outcomes, it is essential to focus on better access to early diagnosis, treatment, and support for individuals with SCD. By addressing these issues, Africa can significantly improve the lives of those affected and reduce the impact of SCD on the continent.
1. Williams TN. Sickle Cell Disease in Sub-Saharan Africa. Hematol Oncol Clin North Am. 2016 Apr;30(2):343-58. doi: 10.1016/j.hoc.2015.11.005. Available from here.
2. World Health Organization Africa Regional. Sickle Cell Disease in the African Region. [Internet]. 22 June 2010. [Cited January 15, 2025. Available from here.
3. World Health Organization. Sickle Cell Disease. [Internet]. 6 August 2025. [Cited December 15, 2025]. Available from here.
4. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi: 10.1016/s0140-6736(10)61029. Abstract available from here.
5. Serjeant, GR. The natural history of sickle cell disease. Cold Spring Harbor Perspectives in Medicine, 2013;3(10), a011783. Available from here.
6. Weatherall DJ, Clegg JB. Inherited hemoglobin disorders: An increasing global health problem. Bulletin of the World Health Organization, 2001;79(8), 704–712. Abstract available from here.
7. Tshilolo L, Kafando E, Sawadogo M, Cotton F, Vertongen F, Ferster A, Gulbis B. Neonatal screening and clinical care programmes for sickle cell disorders in sub-Saharan Africa: Lessons from pilot studies, Public Health, 2008;122(9)933- doi: 10.1016/j.puhe.2007.12.005. Available from here.
Published: January 28, 2025
Updated: December 15, 2026
© 2025. Datelinehealth Africa Inc. All rights reserved.
Permission is given to copy, use and share content freely for non-commercial purposes without alteration or modification and subject to source attribution
DATELINEHEALTH AFRICA INC., is a digital publisher for informational and educational purposes and does not offer personal medical care and advice. If you have a medical problem needing routine or emergency attention, call your doctor or local emergency services immediately, or visit the nearest emergency room or the nearest hospital. You should consult your professional healthcare provider before starting any nutrition, diet, exercise, fitness, medical or wellness program mentioned or referenced in the DatelinehealthAfrica website. Click here for more disclaimer notice.


